
Historie:
- 1994: In der Medizintechnik wird das Qualitätsmanagement mit der ISO 9001 normativ eingeführt.
- Etwa zur gleichen Zeit wurde von der FDA erstmals ein regulierter, eigenständiger Bereich etabliert. Durch diesen wurde beispielsweise die „Quality System Regulation“ herausgegeben, in welcher zum ersten Mal die Anforderungen an ein System zum Qualitätsmanagement für medizinische Produkte festgeschrieben wurden.
- Unter Rekurs auf diese Entwicklung sahen sich auch die Europäer dazu genötigt, eine eigenständige Qualitätsmanagement-Norm für medizinische Produkte zu erarbeiten.
- 2003: Erste eigenständige Qualitätsmanagement-Norm für Medizinprodukte in Europa tritt in Kraft.
- Schließlich wurde eine zur Harmonisierung vorgesehene Übergangszeit von drei Jahren festgelegt.
- Seit deren Ende im Jahr 2006 blieb zumindest der normative Teil dieser zweiten Ausgabe bis zum Jahr 2015 unverändert.
- 2014: Fertigstellung der Erstfassung nach der Überarbeitung, 2015: Freigabe der neuen Fassung ISO 9001: 2015.
QMS in der Medizintechnik
- Nachweis der Qualität eines medizinischen Produktes wird von immer mehr Kunden erwartet.
- So empfinden es viele Kunden nicht mehr als ausreichend, wenn eine reine Qualitätskontrolle zum Beispiel im Rahmen einer Endkontrolle durch den Hersteller durchgeführt wird.
- Die Aufgabe des Qualitätsmanagementsystems (QMS) ist es, die Qualitätsanforderungen über den gesamten Produktionszyklus hinweg zu überwachen – beginnend bei der Materialzulieferung bis zur Produktauslieferung.
- Durch das QMS werden auf der einen Seite die Qualitätsziele definiert und auf der anderen Seite festgelegt, mithilfe welcher Maßnahme die jeweiligen Qualitätsziele erreicht werden sollen.
- Der Beschluss des QMS erfolgt von der Leitung eines Unternehmens und die Umsetzung von den jeweiligen Fachabteilungen.
- Laut der DIN EN ISO 9001: 2008 kommt dem Qualitätsmanagementsystem die Aufgabe zu, Leitung und Lenkung einer Organisation in Bezug auf die Qualität zu gewährleisten.
- Anders formuliert: Durch ein Qualitätsmanagementsystem ist es möglich, Prozesse so zu konzipieren, dass das Medizinprodukt den Kundenanforderungen in Bezug auf die Qualität entspricht. Zudem kann sichergestellt werden, dass auch es auch den rechtlichen Anforderungen genügt.
- Diese lassen sich folgendermaßen zusammenfassen:
- Das Medizinprodukt sollte von seiner Qualität so beschaffen sein, dass es seine Zweckbestimmung möglichst fehlerfrei erfüllt.
- Das Medizinprodukt sollte eine Qualität aufweisen, die sicherstellt, dass die sich beim Gebrauch des Medizinprodukts ergebenden Risiken sowohl für den Menschen als auch für die Umwelt vertretbar sind.
- Dabei bezieht sich das Qualitätsmanagement in der Medizintechnik vorrangig auf produkt- und kundebezogene Prozesse im Unternehmen. Bei diesen handelt es sich unter anderem um Beschaffung, Entwicklung, Produktion, Vertrieb, Marketing, Auslieferung, Installation bzw. Montage, Instandhaltung sowie den Kundendienst.
Ziele des QMS
- Ein Qualitätsmanagementsystem kann sich auf zahlreiche Leistungen und Produkte beziehen.
- Aufgrund der Tatsache, dass Medizinprodukte am Menschen eingesetzt werden, müssen sie hohe Erwartungen in Bezug auf die Zuverlässigkeit und Sicherheit erfüllen.
- Aufgrund von Verordnungen und Gesetzen wird durch den Gesetzgeber für einen hohen Schutz für Patienten, Anwender sowie Dritte gesorgt.
- Deshalb muss ein Medizinprodukt alle beschriebenen Leistungen ordnungsgemäß und zuverlässig erfüllen. Nur dann genügt das Medizinprodukt den hohen Sicherheitskriterien und kann den Gesundheitsschutz sicherstellen.
- Medizinprodukt-Hersteller müssen alle Gefahrenpotenziale mithilfe des aktuellen Stands der Technik soweit reduzieren wie möglich.
- Zur Sicherung der Qualität im Rahmen der Produkthaftung ist aus diesem Grund ein Qualitätsmanagementsystem unverzichtbar.
- Die Implementierung eines Qualitätsmanagementsystems ist für Unternehmen mit enormen finanziellen und zeitlichen Ressourcen verbunden, allerdings ergeben sich auf lange Sicht Einsparungen. Denn durch die Einführung eines Qualitätsmanagementsystems können Entwicklungszeiten verkürzt, Prozesse optimiert und Fehler vermieden werden.
Rechtliche Grundlagen
- Aufgrund der oben erörterten Umstände schreibt die noch bis zum Ende der Übergangsfrist für die neue MDR anwendbare Richtlinie 93/42/EWG für Medizinprodukte (MDD), die spezifischen Risikoklassen zugehören, ein Qualitätsmanagementsystem vor.
- Dieses ist dann bei der Entwicklung über die Herstellung bis hin zum Inverkehrbringen des jeweiligen medizinischen Produktes anzuwenden.
- Im Rahmen der Richtlinie wird in der Medizintechnik diesbezüglich zwischen drei Kategorien von Systemen zum Qualitätsmanagement differenziert:
- Vollständiges System zum Qualitätsmanagement: In dieser Kategorie wird das Qualitätssicherungssystem sowohl für die Auslegung als auch die Fertigung sowie die Endkontrolle des jeweiligen Medizinproduktes eingesetzt.
- Qualitätssicherung der Produktion: Das System zum Qualitätsmanagement dient zur Qualitätssicherung und ist einerseits für die Herstellung und andererseits für die Qualitätsprüfung bzw. Endkontrolle der jeweiligen medizinischen Produkte vorgesehen.
- Qualitätssicherung des (Medizin-)Produkts: Das System zum Qualitätsmanagement ist für die Endkontrolle sowie die Prüfung des jeweiligen Medizinprodukts vorgeschrieben.
- Hinzu kommt, dass in Abhängigkeit des vorgesehenen Konformitätsbewertungsverfahrens oder der Risikoklasse eine Zertifizierung des Qualitätsmanagementsystems in der Medizintechnik zwingend notwendig sein kann.
- Je nachdem, in welchen Ländern ein Medizinprodukt vermarktet werden soll, muss der Hersteller das Qualitätsmanagementsystem so konzipieren, dass neben den rechtlichen Anforderungen der EU auch diejenigen der entsprechend anvisierten Länder berücksichtigt.
- Dies ist zum Beispiel dann der Fall, wenn der Hersteller für das Medizinprodukt eine Zulassung der FDA anstrebt.
- Sowohl für den US-amerikanischen als auch für den europäischen Markt gelten dabei Richtlinien, die der Qualitätssicherung nach GMP (Good Manufacturing Practice), das heißt der guten Herstellungspraxis, entsprechen. Hinzu kommt, dass sich Hersteller medizinischer Produkte über die Qualitätsmanagementsysteme seiner Zulieferer informieren müssen. Denn auch für zugekaufte Produktkomponenten gilt es in der Medizintechnik, Qualitätsnachweise zu erbringen.
- Im Allgemeinen liegt dem Zertifizierungsprozess eine Qualifizierung bzw. Validierung zugrunde. Diese wird unter Rekurs auf das Lebenszyklusmodell in die folgenden Phasen unterteilt:
- Designqualifizierung (DQ, Design Qualification)
- Installationsqualifizierung (IQ, Installation Qualification)
- Funktionsqualifizierung (OQ, Operational Qualification)
- Leistungsqualifizierung (PQ, Performance Qualification)
- Unterstützung diesbezüglich bietet beispielsweise der Gemeinsame Bundesausschuss G-BA, welcher die Ergebnisse der Qualitätssicherung nach § 137 SGB V vom durchführenden Institut veröffentlichen lässt.
Zertifizierung eines Qualitätsmanagement-Systems – Wie erhalte ich das QMS Zertifikat?
- Um ein Qualitätsmanagementsystem zertifizieren zu lassen, muss ein Antrag auf eine Qualitätsmanagement-Zertifizierung bei einer Benannten Stelle gestellt werden. Im Anschluss wird ein zweistufiges Auditverfahren durchgeführt, das heißt ein Auditor überprüft, ob die Vorgaben vom Unternehmen eingehalten werden.
- Darüber hinaus wird auch das Qualitätsmanagement der technischen Dokumentation des Medizinprodukts überprüft.
- Hersteller von medizinischen Produkten können diese einerseits nach der DIN EN ISO 9001 und andererseits nach der DIN EN ISO 13485 zertifizieren lassen.
- Um die Qualität in der Medizintechnik sicherzustellen ist für Medizinprodukte die DIN EN ISO 13485 „Medizinprodukte-Qualitätsmanagementsysteme – Anforderungen für regulatorische Zwecke“ in der Regel die bedeutsamere Option.
- In dieser Norm werden die Anforderungen an das Qualitätsmanagementsystem des Herstellers der Medizinprodukte sowie die damit einhergehenden Dienstleistungen definiert.
- Durch Anwendung der Norm soll sichergestellt werden, dass Herstellung und Verpackung von medizinischen Produkten und die aus ihrem Gebrauch entstehenden Risiken sowohl für den Menschen als auch die Umwelt einen vertretbaren Rahmen nicht überschreiten.
- Aus diesem Grund können Medizinproduktehersteller, welche ein Qualitätsmanagementsystem entsprechend der DIN EN ISO 13485 nachweisen, zugleich belegen, dass ihr medizinisches Produkt den grundlegenden Anforderungen in Bezug auf die Qualitätssicherung bzw. die Qualität genügt. Auch die Qualitätssicherung der Messtechnik, Qualitätssicherung der Werkzeuge und Qualitätssicherung der Fertigung fallen ebenso unter diese Kategorie, wie die Qualitätssicherung im Wareneingang (Wareneingangskontrolle) und die Qualitätssicherung im Warenausgang.
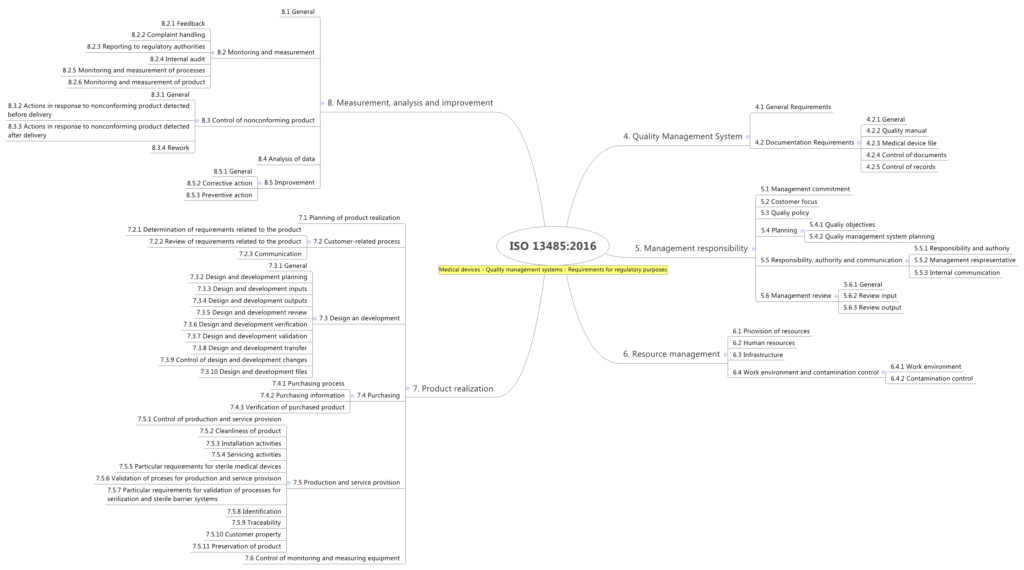
Aufbau der ISO 13485
Abschnitt 4 (Einleitung)
Effektives Qualitätsmanagementsystem (QMS)
- Definiert Prozesse, aus denen sich das QMS zusammensetzt und zeigt, wie sie mit einem risikobasierten Ansatz verbunden und gesteuert werden.
- Qualitätsmanagementhandbuch
- Medical Device File für jedes Medizinprodukt
- Dokumente und Aufzeichnungen müssen entsprechend einem dokumentierten Prozess gesteuert werden
Abschnitt 5 (Verantwortung der Leitung)
Verpflichtung des Top-Managements für den Aufbau und die Aufrechterhaltung eines effektiven Qualitätsmanagementsystems
- Qualitätspolitik: Festlegung von Qualitätszielen für die gesamte Organisation
- Festlegung von Verantwortlichkeiten und Zuständigkeiten für den effektiven Betrieb der Organisation in Übereinstimmung mit dem Qualitätsmanagementsystem
- Managementbeauftragte, die die Effektivität durch Management Reviews belegen
Abschnitt 6 (Management von Ressourcen)
Arbeitsumgebung und Personal
- Die Leute müssen sich auskennen (Schulung, Weiterbildung, Qualifikation)
- Arbeitsumgebung
- Gesundheit
- Sauberkeit
- Kontamination (wenn Zutreffend)
Abschnitt 7 (Produktrealisierung)
Betrifft quasi den gesamten Betrieb
- Planung der für die Produktrealisierung notwendigen Prozesse
- Qualitätsziele = Anforderungen an die Produktrealisierung definieren
- Vorkehrungen für die Kommunikation mit Kunden und Aufsichtsbehörden für Medizinprodukte treffen
- Organisation von Design- und Entwicklungsprozessen
- Designverifizierung und -validierung
- Designtransfer (in die Fertigung)
- Verfahren zur Steuerung des Einkaufs von Produkten und Dienstleistungen
- Auswahl der Lieferanten und Überwachung ihrer Leistung
- Produktkäufe planen und überprüfen, ob die gekauften Produkte den Spezifikationen entsprechen.
- Kontrolle und Überprüfung der gekauften Produkte
- Planung, Überwachung und Steuerung der Service- und Produktionsbereitstellung
- Kontaminationskontrolle (falls zutreffend)
- Installations- und Verifikationsanforderungen
- Serviceverfahren und Referenzmaterialien
- Validierung von Produktionsprozessen, bei denen eine Inspektion nicht möglich ist.
- Produktidentifikation und Rückverfolgbarkeit
Abschnitt 8 (Messung, Analyse und Verbesserung)
Planen, wie die Organisation Prozesse überwachen, messen und analysieren wird, um die Konformität und Effektivität von Produkten und QMS sicherzustellen
- Verfahren zur Einholung und Überwachung von Kundenfeedback
- Verfahren zur Untersuchung von Beanstandungen
- Verfahren zur Überprüfung von Risiken
- Verfahren zur Meldung an Regulierungsbehörden
- Planen und durchführen interner Audits, um festzustellen, ob das QMS konform ist und die Prozesse die geplanten Ergebnisse erreichen
- Überwachung und Messung der Produkte während des Herstellungsprozesses, um nicht konforme Produkte zu identifizieren
- geeignete Maßnahmen ergreifen, um nicht konforme Produkte zu isolieren
- unbeabsichtigte Lieferung oder Verwendung von nicht konformen Produkten verhindern
- Daten über das QMS der Organisation auswerten
- Bewertung der Eignung und Wirksamkeit des QMS
- Verbesserungen durchführen, wo erforderlich/identifiziert
- gegebenenfalls Korrektur- und Vorbeugemaßnahmen ergreifen
-
- ISO 13485_2016_EN
-
- QMS
Qualitätsmanagementshandbuch
Was ist ein QM-Handbuch?
- Zentraler Startpunkt zum QM-System
- Qualitätspolitik des Unternehmens erklären
- Übersicht über den Aufbau des QM-Systems
- Zusammenspiel der Prozesse
Was steht in einem QM-Handbuch?
- Philosophie und Ziele
- Visionen und Strategien, die die Firma ausmachen
- Kein allgemeines gerede
- Qualitätspolitik
- Ausgehend von der Philosophie und den Zielen eine Politik beschreiben. Beispielsweise:
- Eine große Anzahl günstiger Produkte an möglichst viele Menschen verkaufen?
- Patientensicherheit ist immer an erster Stelle
- Gebrauchstauglichkeit ist immer an erster Stelle
- Qualitätsziele
- Ausgehend von der Qualitätspolitik können Qualitätsziele abgeleitet werden.
- Jedes Qualitätsziel sollte eine Metrik beinhalten.
- Ziele sollten SMART formuliert sein. Wobei die Zeitliche Terminierung im QM-Zielen nicht immer sinnvoll ist…
- Produkte sollen mindestens 10% günstiger sein, als die von der Konkurrenz
- Bei Umfragen unserer Produkte soll die Bedienung um mindestens 20% einfacher sein, als die der Konkurrenz
- Wir wollen einem weltweiten Marktanteil von mindestens 65% im Bereich der professionellen Dialysemaschinen
- Die Produkten sollen so sicher sein, dass 10% weniger Mitteilungen an die BfarM gemeldet werden müssen als bei Konkurenzprodukten
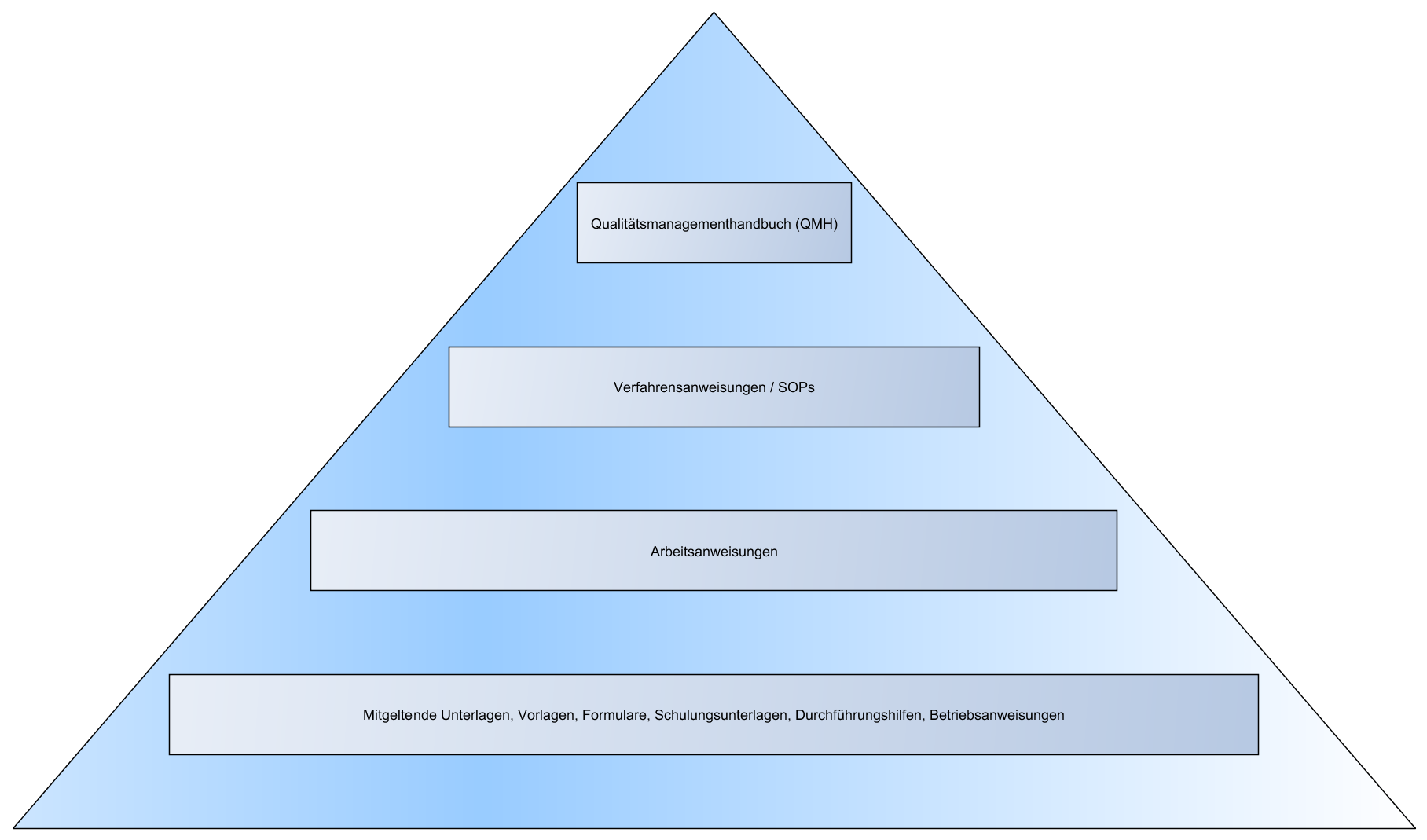
- Organigramm
- Struktur des QM-Systems
- „Dokumentenpyramide“
- Rollenbeschreibungen (für’s QM-System relevant)
- z.B. Importeuer, Hersteller, …
- Prozesse
- Übersicht und Zusammenhänge
- Kernprozesse
Qualitätsmanagement funktioniert top-down. Also von der Führungsebene nach unten!
Wenn das QM-Handbuch nicht gelesen wird, kann das Rückschlüsse auf die Führungsebene zulassen…
Quelle: https://www.johner-institut.de/blog/qualitaetsmanagement-iso-13485/
Umgang mit Dokumenten und Aufzeichnungen
Dokumente
Dokumente haben einen Vorgabecharakter. Sie beinhalten meist Beschreibungen für etwas (Prozess, Produkt, …) oder enthalten Forderungen (Tätigkeit, Produkt, …). Ein Dokument ist veränderbar. Das bedeutet, es können verschiedene Versionsstände eines Dokumentes existieren. Die Dokumente wachsen quasi mit den Unternehmen mit.
Beispiele:
- Qualitätsmanagementhandbuch
- Verfahrensanweisungen
- Projektpläne, Entwicklungspläne
- Anforderungsokumente eines Produktes
- Architekturdokumente eines Produktes
- …
Anforderungen an Dokumente
- Vor Veröffentlichung/Herausgabe/Verteilung bewerten und genehmigen
- Bewerten und wenn nötig erneut aktualisieren und erneut genehmigen
- Sicherstellen, dass Änderungen erkennbar sind
- Sicherstellen, dass stets die aktuelle Version verfügbar ist
- Lesbarkeit sicherstellen
- Sicherstellen, dass externe Dokumente identifiziert und gelenkt werden
- Sicherstellen, dass ein Verlust verhindert wird
- Sicherstellen, dass veraltete Dokumente nicht mehr verwendet werden
Aufzeichnungen:
Aufzeichnungen haben einen Nachweischarakter. Sie dienen als Nachweis für etwas (erreichte Ergebnisse, Erfüllung von Anforderungen, Wirksamkeit eines Prozesses). Eine Aufzeichnung ist nach der Erstellung nicht veränderbar ist. Sie zeichnen quasi einen bestimmten Zustand zu einem festen Zeitpunkt auf. Da Aufzeichnungen nicht veränderbar sind, gibt es auch keine Versionsstände.
Beispiele:
- (Prüf)Protokolle
- (Audit)Prüfberichte
- Managementbewertungen
- …
Anforderungen an Aufzeichnungen
- Dokumentation von Lagerung und Schutz
- Hierzu muss im ein Verfahren im QMS hinterlegt sein
- Lesbarkeit und Auffindbarkeit sicherstellen
- Aufzeichnungen müssen mindestens über die Lebensdauer des Medizinproduktes aufbewahrt werden
- Jedoch mindestens zwei Jahre
- Anwendbare regulatorische Anforderungen müssen erfüllt werden (z.B. gesetzliche Anforderungen an die Aufbewahrung)
Was dann doch häufig schief geht
- Die Mitarbeiter wissen nicht wie sie auf das QM-System zugreifen können
- Veraltete Dokumente befinden sich in Umlauf
- Die Dokumente der technischen Dokumentation eines Produktes wurden nicht aktualisiert
- Die Chronologie der Dokumentenfreigabe macht keinen Sinn
- z.B. Testergebnisse (Aufzeichnung) wurden vor den Anforderungsdokumenten freigegeben
- Der stand eines Dokumentes ist nicht ersichtlich (Entwurf, Freigegeben, Zurückgezogen)
- Schulungsnachweise für geänderte Dokumente sind nicht nachweisbar
Mögliche technische Umsetzungen
Je nach Unternehmen werden Dokumente und Aufzeichnungen
- Mit Wiki-Systemen (mediawiki, confluence, …)
- Mit Office-Programmen (Dateisystem, Sharepoint, Versionsverwaltung, …)
- Mit speziellen Tools (Polarion, IBM Doors, CodeBeamer, ocranos, ttc integrity, Aligned Elements, greenlight guru GO and GROW, …)
(Wichtige) Zu dokumentierende Verfahren
Liste der zu dokumentierenden Verfahren:
- Lenkung von Dokumenten
- Lenkung von Aufzeichnungen
- Managementbewertung
- Fähigkeiten, Bewusstsein und Schulungen
- Arbeitsumgebung
- Risikomanagement
- Design und Entwicklung
- Einkauf und Beschaffung
- Sauberkeit
- Installation, Service und Instandhaltung
- Validierung von Computer-Software
- Sterilisierung (wenn Anwendbar)
- Produktidentifikation (UDI, kommt mit der MDR)
- Rückverfolgbarkeit
- Produkterhaltung
- Lenkung von Überwachungs- und Messmitteln
- Rückmeldungen
- Lenkung von Entwicklungsänderungen
- Kundenrückmeldungen
- Interne Audits
- Überwachung und Messung von Prozessen und Produkten
- Umgang mit nichtkonformen Produkten
- Datenanalyse
- Korrektur- und Vorbeugemaßnahmen (CAPA)
Die Liste erhebt nicht den Anspruch auf Vollständigkeit, sondern dient dazu den Umfang zu zeigen was ein ISO 13485 zertifiziertes Unternehmen alles dokumentieren muss.
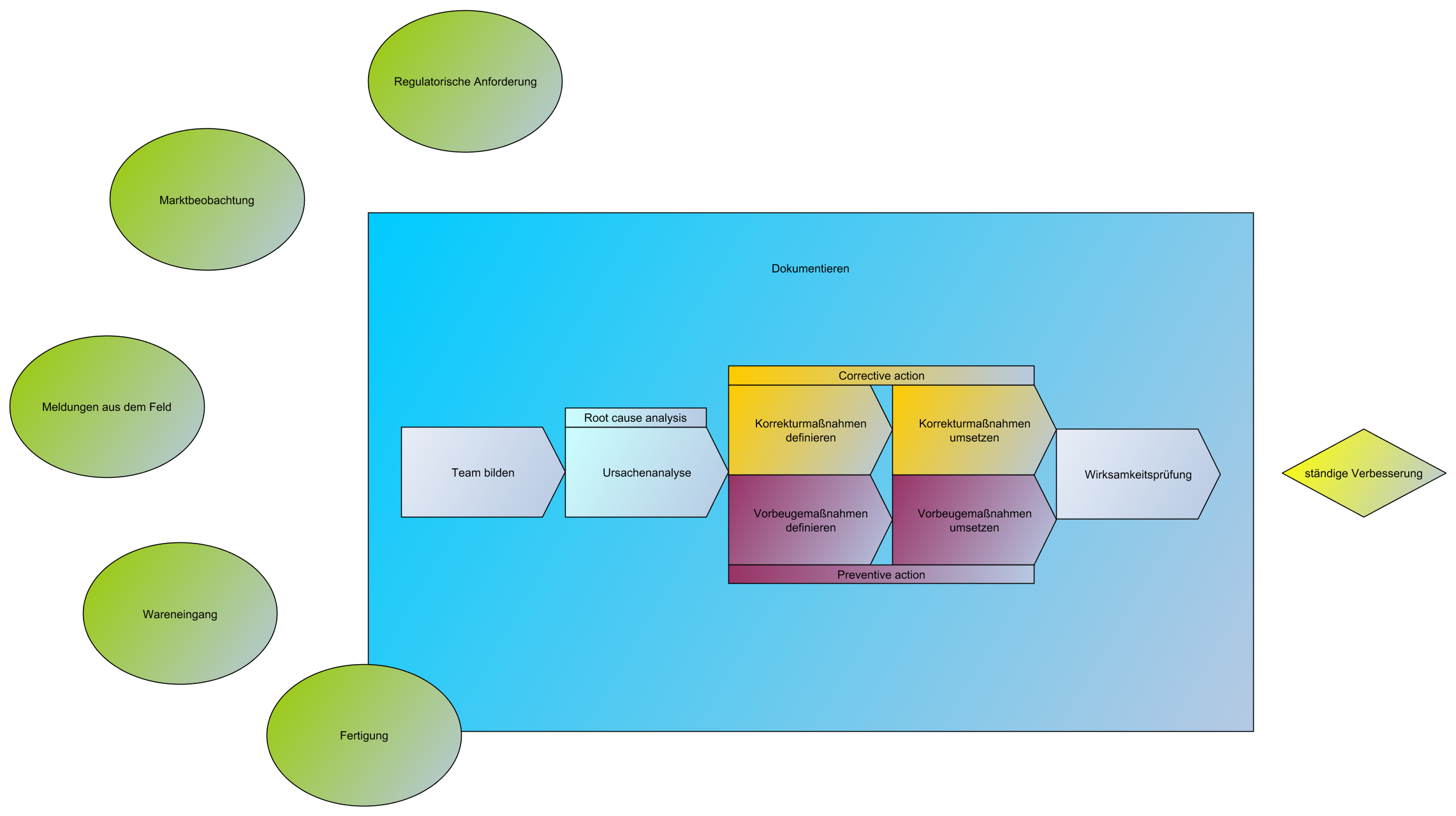
Ständige Verbesserung / Korrektur- und Vorbeugemaßnahmen / CAPA
- Der Hersteller muss jederzeit konforme Produkte im Markt haben.
- Die Sicherheit für Patienten, Anwender und Dritte ist das höchste Ziel.
- Deshalb muss eine Marktbeobachtung durchgeführt werden (post market surveillance)
- Im Falle eines Fehlers/Abweichung muss zum einen der Fehler selbst, als auch das Wiederauftreten des Fehlers behoben/vermieden werden
- Dann gibt es eine Ursachenanalyse (root cause analysis), die ausreichend tief zu hinterfragen ist, wie es dazu kam.
- Bsp: Fahrrad hat nen Platten –> Tägliches Flicken –> Ursache: Nägel im Hof
- Danach kommt die Korrekturmaßnahme (Corrective Action):
- Das Ziel der Korrekturmaßnahme besteht also darin, nicht nur Fehler, sondern die Ursachen von bereits aufgetretenen Fehlern zu erkennen und zu beseitigen und sicherzustellen, dass solche Fehler nicht nochmal auftreten.
- Bsp: Nägel im Hof zusammenkehren
- Dann folgt die Vorbeugemaßnahme (Preventive Action):
- Die Vorbeugemaßnahmen sollen künftige Fehler vermeiden, die noch nicht aufgetreten sind.
- Bsp: Regelmäßige Kehrordnung im Hof einführen
Typische CAPAs sind:
- Rückrufe
- Major findings bei externen Audits
- kritische Abweichungen bei internen Audits
- Umstände, welche die Konformität des Produktes beeinflussen etc.
-
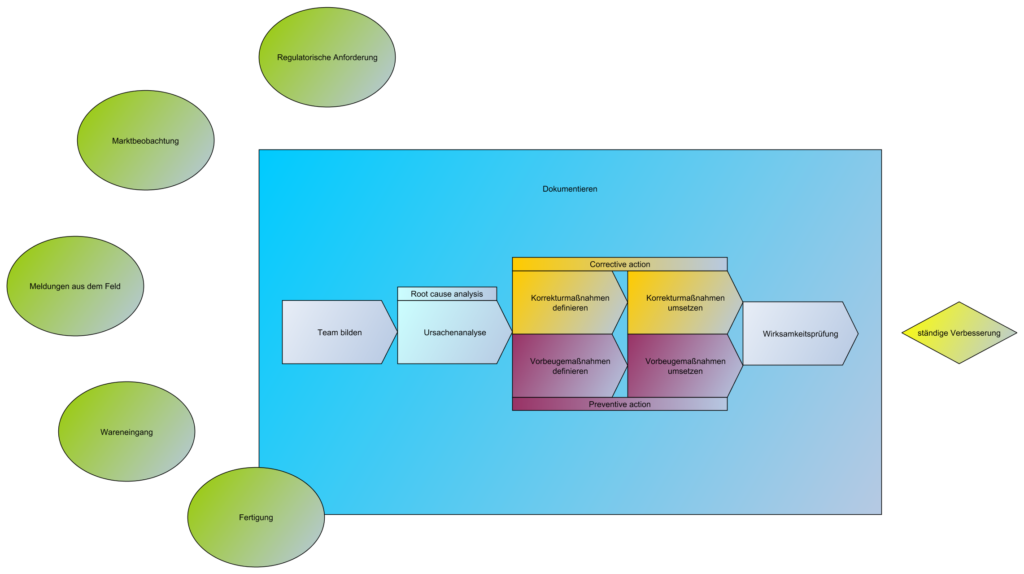
- CAPA
weitere Quelle:
- https://www.bvmed.de/de/bvmed/presse/pressemeldungen/medinform-veranstaltung-zum-qualitaetsmanagement-fuer-medizinprodukte-die-neue-norm-en-iso-13485-ist-prozessorientiert
- https://www.johner-institut.de/blog/regulatory-affairs/dokumentenlenkung/
- https://www.germersdorf-beratung.de/2018/01/08/die-neue-iso-13485-2016-ein-%C3%BCberblick/
- https://www.devicemed.de/qualitaetsmanagement-in-der-medizintechnik-definitionen-ziele-und-tools-a-710821/
- https://www.qz-online.de/qualitaets-management/qm-basics/recht_normen/branchenspezifische_anforderungen_qm_systeme/artikel/iso-13485-2016-was-hat-sich-geaendert-1629193.html
- https://medtech-ingenieur.de/iso-13485-was-steht-da-eigentlich-drin/

Schreibe einen Kommentar